File / Class Conversion
Examples are given for conversion between CRYSTALpytools classes and external classes. Functions tested here are defined in the convert module.
CRYSTAL <-> pymatgen
The ‘cry_out2pmg()’ function
cry_out2pmg() converts a CRYSTAL output file into a pymatgen Structure/Molecule object, which is different from the crystal_io.Crystal_ouput.get_geometry() method, where the geometry.CStructure class is returned.
Get the optimized geometry of MgO.
[1]:
from CRYSTALpytools.convert import cry_out2pmg
struc = cry_out2pmg('conv_mgoOpt.out',initial=False)
print(struc)
Full Formula (Mg1 O1)
Reduced Formula: MgO
abc : 2.998288 2.998288 2.998288
angles: 60.000000 60.000000 60.000000
pbc : True True True
Sites (2)
# SP a b c
--- ---- ---- ---- ----
0 Mg 0 0 0
1 O -0.5 -0.5 -0.5
Get a Molecule class for CO.
[2]:
from CRYSTALpytools.convert import cry_out2pmg
struc = cry_out2pmg('conv_coOpt.out',initial=False, molecule=True)
print(struc)
Full Formula (C1 O1)
Reduced Formula: CO
Charge = 0.0, Spin Mult = 1
Sites (2)
0 C -0.063726 -0.039829 -0.031863
1 O 0.863726 0.539829 0.431863
The ‘cry_gui2pmg’ function
cry_gui2pmg() converts a CRYSTAL GUI (fort.34) file into a pymatgen Structure/Molecule object.
NOTE
Currently, symmetry information of GUI class is lost during conversion.
[3]:
from CRYSTALpytools.convert import cry_gui2pmg
struc = cry_gui2pmg('conv_paracetamol.gui')
print(struc)
Full Formula (H36 C32 N4 O8)
Reduced Formula: H9C8NO2
abc : 7.161425 9.265602 12.677166
angles: 90.000000 116.348749 90.000000
pbc : True True True
Sites (80)
# SP a b c
--- ---- --------- --------- ---------
0 O 0.05598 0.43021 -0.286803
1 O -0.05598 -0.06979 -0.213197
2 O -0.05598 -0.43021 0.286803
3 O 0.05598 0.06979 0.213197
4 O 0.453896 -0.002239 0.302393
5 O 0.546104 0.497761 0.197607
6 O -0.453896 0.002239 -0.302393
7 O -0.546104 -0.497761 -0.197607
8 N -0.212129 0.043273 0.339866
9 N 0.212129 -0.456727 0.160134
10 N 0.212129 -0.043273 -0.339866
11 N -0.212129 0.456727 -0.160134
12 C -0.149616 0.137894 0.436285
13 C 0.149616 -0.362106 0.063715
14 C 0.149616 -0.137894 -0.436285
15 C -0.149616 0.362106 -0.063715
16 C -0.237476 0.141661 -0.485502
17 C 0.237476 -0.358339 -0.014498
18 C 0.237476 -0.141661 0.485502
19 C -0.237476 0.358339 0.014498
20 C -0.166071 0.239609 -0.393174
21 C 0.166071 -0.260391 -0.106826
22 C 0.166071 -0.239609 0.393174
23 C -0.166071 0.260391 0.106826
24 C -0.004707 0.33468 -0.376123
25 C 0.004707 -0.16532 -0.123877
26 C 0.004707 -0.33468 0.376123
27 C -0.004707 0.16532 0.123877
28 C 0.086433 0.328413 -0.452801
29 C -0.086433 -0.171587 -0.047199
30 C -0.086433 -0.328413 0.452801
31 C 0.086433 0.171587 0.047199
32 C 0.014023 0.231484 0.4547
33 C -0.014023 -0.268516 0.0453
34 C -0.014023 -0.231484 -0.4547
35 C 0.014023 0.268516 -0.0453
36 C 0.600808 -0.017228 0.277576
37 C 0.399192 0.482772 0.222424
38 C -0.600808 0.017228 -0.277576
39 C -0.399192 -0.482772 -0.222424
40 C 0.581296 -0.106752 0.174364
41 C 0.418704 0.393248 0.325636
42 C -0.581296 0.106752 -0.174364
43 C -0.418704 -0.393248 -0.325636
44 H -0.361418 0.069092 -0.494845
45 H 0.361418 -0.430908 -0.005155
46 H 0.361418 -0.069092 0.494845
47 H -0.361418 0.430908 0.005155
48 H -0.237356 0.240664 -0.333637
49 H 0.237356 -0.259336 -0.166363
50 H 0.237356 -0.240664 0.333637
51 H -0.237356 0.259336 0.166363
52 H -0.787752 0.400762 -0.441974
53 H -0.212248 -0.099238 -0.058026
54 H 0.787752 -0.400762 0.441974
55 H 0.212248 0.099238 0.058026
56 H 0.086988 0.228584 0.396029
57 H -0.086988 -0.271416 0.103971
58 H -0.086988 -0.228584 -0.396029
59 H 0.086988 0.271416 -0.103971
60 H 0.195544 0.469876 -0.265796
61 H -0.195544 -0.030124 -0.234204
62 H -0.195544 -0.469876 0.265796
63 H 0.195544 0.030124 0.234204
64 H -0.105395 0.028427 0.308708
65 H 0.105395 -0.471573 0.191292
66 H 0.105395 -0.028427 -0.308708
67 H -0.105395 0.471573 -0.191292
68 H 0.433006 -0.085398 0.098483
69 H 0.566994 0.414602 0.401517
70 H -0.433006 0.085398 -0.098483
71 H -0.566994 -0.414602 -0.401517
72 H -0.292847 -0.088761 0.149936
73 H 0.292847 0.411239 0.350064
74 H 0.292847 0.088761 -0.149936
75 H -0.292847 -0.411239 -0.350064
76 H 0.580205 -0.220848 0.194754
77 H 0.419795 0.279152 0.305246
78 H -0.580205 0.220848 -0.194754
79 H -0.419795 -0.279152 -0.305246
With the vacuum option, vacuum layer with the specified thickness is inserted along the non-periodic direction(s). The returned structure is 3D periodic. For non-periodic structures, set vacuum=None.
[4]:
from CRYSTALpytools.convert import cry_gui2pmg
struc = cry_gui2pmg('conv_mgo100.gui', vacuum=20)
print(struc)
Full Formula (Mg2 O4)
Reduced Formula: MgO2
abc : 3.009789 3.009789 26.128240
angles: 90.000000 90.000000 90.000000
pbc : True True True
Sites (6)
# SP a b c
--- ---- --- --- ---------
0 Mg 0 0.5 0.040727
1 Mg 0.5 0 -0.040727
2 O 0 0.5 -0.040727
3 O 0.5 0 0.040727
4 O 0.5 0 -0.117272
5 O 0 0.5 0.117272
Alternatively, a 2D periodic structure is returned with default 500 Angstrom along non-periodic direction(s).
[5]:
from CRYSTALpytools.convert import cry_gui2pmg
struc = cry_gui2pmg('conv_mgo100.gui')
print(struc)
Full Formula (Mg2 O4)
Reduced Formula: MgO2
abc : 3.009789 3.009789 500.000000
angles: 90.000000 90.000000 90.000000
pbc : True True False
Sites (6)
# SP a b c
--- ---- --- --- ---------
0 Mg 0 0.5 0.002128
1 Mg 0.5 0 -0.002128
2 O 0 0.5 -0.002128
3 O 0.5 0 0.002128
4 O 0.5 0 -0.006128
5 O 0 0.5 0.006128
The ‘cry_pmg2gui()’ function
cry_pmg2gui() saves a pymatgen structure/molecule object into a CRYSTAL gui object and (optionally) writes it into a gui (fort.34) file.
Setting gui_file=None (default) to disable gui file output
NOTE
Currently, symmetry analysis (symmetry=True) is limited to 3D and 2D cases.
[6]:
from CRYSTALpytools.crystal_io import Crystal_output
from CRYSTALpytools.convert import cry_pmg2gui
out = Crystal_output('conv_mgoOpt.out')
struc = out.get_geometry(initial=False)
struc.make_supercell([[-1, 1, 1], [1, -1, 1], [1, 1, -1]])
mgo_gui = cry_pmg2gui(struc, gui_file='conv_mgoOpt.gui', symmetry=True)
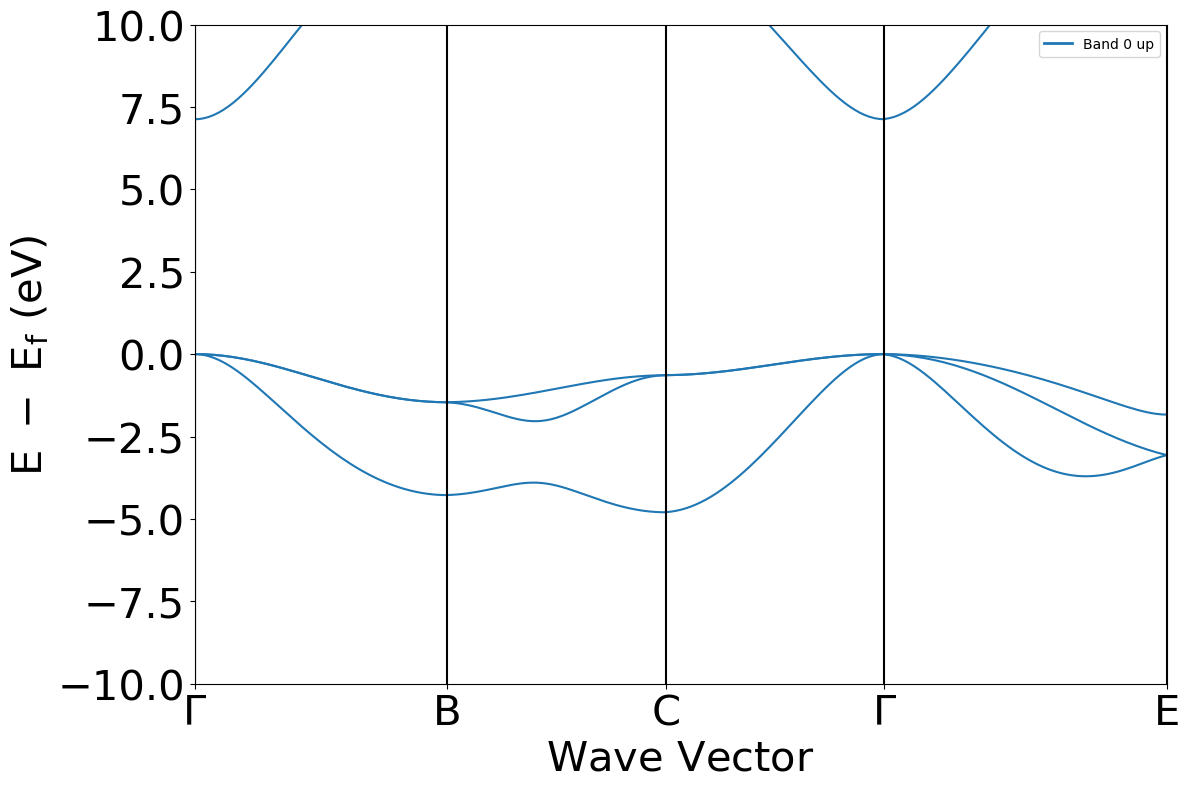
The ‘cry_bands2pmg()’’ function
Essentially this is a short cut for electronics.ElectronBand.to_pmg() method.
Read the band and plot.
[7]:
from CRYSTALpytools.convert import cry_bands2pmg
from pymatgen.electronic_structure.plotter import BSPlotter
bs = cry_bands2pmg('conv_mgoBand.BAND', 'conv_mgoBand.outp',
labels=[r'$\Gamma$','B','C',r'$\Gamma$','E'])
bsplot = BSPlotter(bs)
bsplot.get_plot(ylim=(-10, 10), zero_to_efermi=True)
[7]:
<Axes: xlabel='$\\mathrm{Wave\\ Vector}$', ylabel='$\\mathrm{E\\ -\\ E_f\\ (eV)}$'>
CRYSTAL <-> ASE
Fundamentally, CRYSTALpytools converts its classes to pymatgen classes and calls pymatgen to further convert them into other classes.
The ‘cry_out2ase()’ function
[8]:
from CRYSTALpytools.convert import cry_out2ase
co_ase = cry_out2ase('conv_coOpt.out', initial=True)
print(co_ase)
MSONAtoms(symbols='CO', pbc=False)
The ‘cry_gui2ase()’ function
Convert GUI file/class into ASE ‘Atoms’ class.
[9]:
from CRYSTALpytools.convert import cry_gui2ase
mgo_ase = cry_gui2ase('conv_mgo100.gui', vacuum=10)
print(mgo_ase)
MSONAtoms(symbols='Mg2O4', pbc=True, cell=[[3.00978870041, 0.0, 0.0], [-1.84296404903e-16, 3.00978870041, 0.0], [0.0, 0.0, 16.128239999999998]])
The ‘cry_ase2gui()’ function
Setting gui_file=None (default) to disable gui file output
[10]:
from CRYSTALpytools.convert import cry_ase2gui
from ase.build import bulk
copper_ase = bulk('Cu', 'fcc', a=3.6)
copper_gui = cry_ase2gui(copper_ase, gui_file='conv_CuASE.gui', symmetry=True)
Saving structure files (.cif and .xyz)
The ‘cry_out2cif()’ function
Optional arguments can be put at the end, which will be passed to the pymatgen.io.cif.CifWriter object.
By default, symmetrized structure output is enabled with symprec=0.01.
[11]:
from CRYSTALpytools.convert import cry_out2cif
cry_out2cif('conv_mgo100.out', 'conv_mgo100.cif', vacuum=10, initial=True)
! cat conv_mgo100.cif
# generated using pymatgen
data_MgO2
_symmetry_space_group_name_H-M P4/nmm
_cell_length_a 3.00978870
_cell_length_b 3.00978870
_cell_length_c 16.12824000
_cell_angle_alpha 90.00000000
_cell_angle_beta 90.00000000
_cell_angle_gamma 90.00000000
_symmetry_Int_Tables_number 129
_chemical_formula_structural MgO2
_chemical_formula_sum 'Mg2 O4'
_cell_volume 146.10295240
_cell_formula_units_Z 2
loop_
_symmetry_equiv_pos_site_id
_symmetry_equiv_pos_as_xyz
1 'x, y, z'
2 '-y+1/2, x+1/2, z'
3 '-x, -y, z'
4 'y+1/2, -x+1/2, z'
5 'x+1/2, -y+1/2, -z'
6 '-y, -x, -z'
7 '-x+1/2, y+1/2, -z'
8 'y, x, -z'
9 '-x+1/2, -y+1/2, -z'
10 'y, -x, -z'
11 'x+1/2, y+1/2, -z'
12 '-y, x, -z'
13 '-x, y, z'
14 'y+1/2, x+1/2, z'
15 'x, -y, z'
16 '-y+1/2, -x+1/2, z'
loop_
_atom_site_type_symbol
_atom_site_label
_atom_site_symmetry_multiplicity
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
_atom_site_occupancy
Mg Mg0 2 0.00000000 0.50000000 0.06597868 1
O O1 2 0.00000000 0.50000000 0.18998477 1
O O2 2 0.00000000 0.50000000 0.93402132 1
The ‘cry_gui2cif()’ function
Optional arguments can be put at the end, which will be passed to the pymatgen.io.cif.CifWriter object.
By default, symmetrized structure output is enabled with symprec=0.01.
[12]:
from CRYSTALpytools.convert import cry_gui2cif
cry_gui2cif('conv_paracetamol.gui', 'conv_paracetamol.cif')
! cat conv_paracetamol.cif
# generated using pymatgen
data_H9C8NO2
_symmetry_space_group_name_H-M P2_1/c
_cell_length_a 7.16142460
_cell_length_b 9.26560241
_cell_length_c 12.67716617
_cell_angle_alpha 90.00000000
_cell_angle_beta 116.34874858
_cell_angle_gamma 90.00000000
_symmetry_Int_Tables_number 14
_chemical_formula_structural H9C8NO2
_chemical_formula_sum 'H36 C32 N4 O8'
_cell_volume 753.80006344
_cell_formula_units_Z 4
loop_
_symmetry_equiv_pos_site_id
_symmetry_equiv_pos_as_xyz
1 'x, y, z'
2 '-x, -y, -z'
3 '-x, y+1/2, -z+1/2'
4 'x, -y+1/2, z+1/2'
loop_
_atom_site_type_symbol
_atom_site_label
_atom_site_symmetry_multiplicity
_atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_z
_atom_site_occupancy
H H0 4 0.08698795 0.22858371 0.39602925 1
H H1 4 0.10539508 0.52842732 0.19129183 1
H H2 4 0.19554393 0.03012373 0.23420412 1
H H3 4 0.21224837 0.09923841 0.05802646 1
H H4 4 0.23735644 0.74066371 0.83363740 1
H H5 4 0.29284689 0.08876147 0.85006363 1
H H6 4 0.36141848 0.56909239 0.99484488 1
H H7 4 0.41979469 0.22084824 0.80524578 1
H H8 4 0.43300640 0.58539849 0.59848256 1
C C9 4 0.00470750 0.66532012 0.37612335 1
C C10 4 0.01402274 0.23148403 0.45470045 1
C C11 4 0.08643290 0.17158686 0.04719886 1
C C12 4 0.14961594 0.63789425 0.06371476 1
C C13 4 0.16607109 0.73960930 0.89317382 1
C C14 4 0.23747598 0.64166052 0.98550157 1
C C15 4 0.39919152 0.01722823 0.72242435 1
C C16 4 0.41870361 0.10675211 0.82563609 1
N N17 4 0.21212918 0.54327289 0.16013400 1
O O18 4 0.05597976 0.06978984 0.21319720 1
O O19 4 0.45389631 0.50223886 0.80239301 1
The ‘cry_gui2xyz()’ function
Optional arguments can be put at the end, which will be passed to the pymatgen.io.xyz.XYZ object.
[13]:
from CRYSTALpytools.convert import cry_gui2xyz
cry_gui2xyz('conv_THF.gui', 'conv_THF.xyz')
!cat conv_THF.xyz
13
H8 C4 O1
C 0.741332 2.117653 1.407777
C 1.893667 3.049791 1.036298
C 3.098124 2.139956 1.256255
O 2.655667 0.819485 0.961776
C 1.238012 0.791402 0.842854
H 0.641873 2.054622 2.492302
H -0.216641 2.420182 0.989685
H 1.821281 3.336523 -0.013719
H 1.934790 3.954421 1.639420
H 3.942117 2.379091 0.609359
H 3.435426 2.182677 2.296453
H 0.965393 0.690877 -0.212497
H 0.859259 -0.076681 1.382035
The ‘cry_out2xyz()’ function
Optional arguments can be put at the end, which will be passed to the pymatgen.io.xyz.XYZ object.
[14]:
from CRYSTALpytools.convert import cry_out2xyz
cry_out2xyz('conv_toluene.out', 'conv_toluene.xyz')
!cat conv_toluene.xyz
15
H8 C7
C -0.984612 0.000163 0.014830
C -0.270650 1.195597 0.011653
C -0.270798 -1.195337 0.010052
C -2.488953 0.000108 -0.002093
C 1.117118 1.198581 -0.001608
C 1.116957 -1.198632 -0.002841
C 1.816634 -0.000078 -0.010182
H -0.809924 2.135277 0.022707
H -0.810214 -2.134972 0.020237
H -2.888568 -0.880617 0.498531
H -2.888737 0.885078 0.490855
H -2.864355 -0.004536 -1.026829
H 1.652616 2.138815 -0.002050
H 1.652252 -2.138983 -0.003587
H 2.898133 -0.000164 -0.017475
For more information please refer to the API documentations.